第三节 分子生物学实验诊断技术
一、核酸杂交技术
检验方法建立的基本要素是特异性和灵敏度,在复杂的物体中,使无法感觉的特定物质进入人类的观察范围。核酸杂交检测技术就是利用核酸碱基严格配对的特异性,核酸标记物的灵敏度而建立的检测核酸结构与功能的方法。该法建立以来已有二十年,目前研究实验室用得多,临床实验室用得较少,除成本高外,关键是操作复杂,重复性差,仅能半定量。成本高可随经济发展而缓和,但其它缺点尚需努力克服。
杂交实验的探针应具有特异性,对应不同的杂交方法靶基因(被检基因)应有一定的纯度与丰度。杂交反应的开始是碰撞,探针浓度高则速率增加,一般32P标记探记探针5-100ng/ml,非放射性探针25-1000ng/ml,原位杂交无论放射还是非放射物标记,一般都需0.1-5.0μg/ml。当探针过量时,杂交速率取决于探针长度,如一个100ng/ml20个核苷酸的合成寡核苷酸探针与1-100pg 1kb长的靶基因杂交,10分钟能达到最大杂交率。而相同条件下2kb的克隆探针需160小时。主要原因是这里所指浓度是重量浓度而非摩尔浓度。长探针因标记量大,小的摩尔浓度已足以达到需要的检测灵敏度。为了促进长探针(>250个核苷酸)的杂交速率,加入杂交促进剂是非常必要的。最常用的是硫酸葡聚糖,使用浓度为5%-10%,浓度过高会增加杂交液粘度。聚乙二醇也作为促进剂,价廉且粘度低,但检测本底太高。另外杂交的温度、盐浓度、甲酰胺浓度可调节杂交分子形成的稳定性。理论选用DNA:DNA杂交温度T=Tm-25℃,此时杂交分子最易形成。但具体实验中影响Tm值的因素很多,一般杂交温度低,形成的杂交分子较稳定。RNA:DNA杂交分子的Tm大10-15℃,RNA:RNA杂交分子的Tm大20-25℃,使得杂交温度提高,此时需调节甲酰胺浓度来调节杂交温度。好的实验条件最终应在实践中摸索建立。
(一)斑点杂交
将少量核酸样品点样在硝酸纤维素滤膜上,80℃烘烤后可牢固地固定在膜上,再用探针进行杂交。尼龙膜,特别是聚偏氟乙烯膜(PVDF)与DNA结合力更高,坚韧、易操作。点样可手工,也可用真空点样品。检测可用放射性探针自显影或非放射性探针显色。可用于DNA或RNA分析。下面以非放射性探针分析DNA为例,结果是显色。
取硝酸纤维素膜和滤纸在2xSSC(NaCl 300mmol/L,柠檬酸钠30mmol/L),pH7.0浸15分钟,平铺滤纸在点样抽滤器上,再铺上硝酸纤维滤膜,真空抽气使点样器减压,膜显出凹面。点样50μl(5-10μgDNA,可直接点血清样品)于凹面,撤去真空,把膜晾干。取滤纸浸0.5mol/l NaOH,1.0mol/l NaCl,把膜平铺于滤纸上20分钟,变性。取滤纸二张分别浸在0.5mol/l Tris-Hcl pH7.5和1.0mol/l Tris-HCl,0.6mol/l NaCl pH7.5,分别依次把膜平铺于滤纸上,各中和15分钟,中和后膜的pH为7.2-7.5。于80℃,30-45分钟烘干。(RNA分析点样缓冲液系统不同,无需变性,中和,余大致相同)用塑料封口机把膜和2.5ml预杂交缓冲液封入塑料袋中,42℃,水浴30分钟。
| 预杂交缓冲液: | 65℃6xSSC | (原液:20xSSPE) |
| 5xDenhardt | (50xDenhardt) | |
| 0.5%SDS | (10%SDS) | |
| 100μg/ml变性鲑精DNA片段 | (1mg/ml) | |
| 42℃增加50%甲酰胺 | (原液),配成20ml. |
50xDenhardt:5g聚蔗糖(Ficoll,400型,Parmacia),5g聚乙烯吡咯烷酮和5g牛血清白蛋白(组份Ⅴ,Sigma)加水至500ml。过滤后-20℃保存。
探针2ml(50-100ng/2ml预杂交缓冲液)于100℃,10分钟,马上置冰浴5分钟(变性)。加入袋封口。42℃水浴过夜。去杂交液(可回收)。以2xSSC,0.1%SDS15ml,50℃5分钟洗二次。0.1SSC,0.1%SDS洗二次。封闭:另取袋封入膜和封闭液(蛋白质50mg/ml,100mmol/l Tris-HCl(pH7.5)/150mmol/LNaCl)2.5ml,37℃,30分钟。去封闭液(可回收)。100mmol/lTris-HCl(pH7.5)/150mmol/L NaCl ,15ml,5分钟洗二次。亲和反应:酶标抗体3ml(酶有碱性磷酸酶、过氧化物酶等,标记物有抗地高辛、生物素等,现以地高辛标碱性磷酸酶为例4μg/3ml 100mmol/L Tris-HCl,150mmol/l MgCl2,10mg/ml牛血清白蛋白),封入袋中37℃,30分钟。100mmol/l Tris-Hcl,100mmol/l NaCl,50mmol/l MgCl2,pH9.5,10ml,1分钟洗一次。取硝基四氮唑兰(NBT)75mg/ml 70%二甲基甲酰胺25μl,4-溴-5-氯-3-吲哚磷酸50mg/ml二甲基甲酰胺20μl(底物)和100mmol/l Tris-HCl,100mmol/l NaCl,50mmol/l MgCl2,pH9.56ml混匀,37℃,避光反应三小时,显色。蒸馏水洗膜,晾干。观察结果。
(二)Southern blot
1975年E.Southern发明了将DNA切开,电泳分离,再变性,印迹到硝酸纤维(Nc)滤膜上,用探针进行杂交,检测DNA的方法。自显影或显色用于DNA分析。以32P标记放射性探针为例,放射自显影观察结果。(图18-6)。
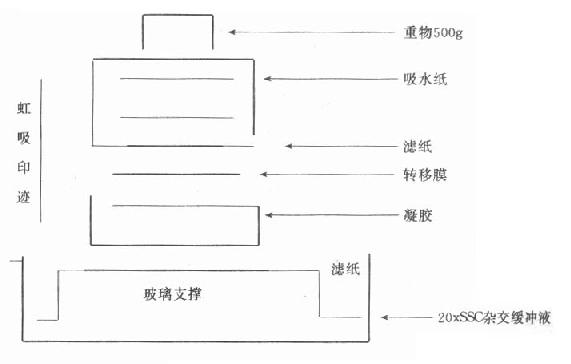
图18-6 Southernblot体系
前述分离、纯化的DNA样品5-10μg/100μlTE,加限制性内切酶3Unit/1μgDNA和内切酶相应缓冲液,在相应温度保温1-3小时(不同的酶所用的缓冲液和温度不同),乙醇沉淀,15-20μl TE溶解DNA断片。前述电泳条件,与DNA分子量标准品(Marker,Ladder)一起,3-4V/cm电泳(30V12-15小时)。在紫外光下观察Marker充分分离,结束电泳。将胶放入0.5mol/l NaCl,0.5mol/L NaOH液体中30分钟,使DNA变性。同时将膜用蒸馏水浸润,在0.5mol/L NaCl,0.5mol/L NaOH液体的方盘皿上,放上滤纸两头浸在液体中,依次放上凝胶、膜、滤纸、一尺厚的吸水纸,压力1kg的重物。转移(印迹,blot)过夜。翌日,把膜放在0.5mol/l Tris-HCl,pH7.0-7.5,1mol/L NaCl液中,中和膜上碱性变性液15-30分钟。戴上手套,用手指压在膜上来回充分洗膜。室温干燥一小时。用塑料封口机,把膜和预杂交缓冲液封在塑料口袋中,注意驱除袋内汽泡。热水浴过夜。探针变性后,加入袋中再封口。热水浴过夜。
| 洗膜: | 2xSSPE,0.5%SDS,总体积200ml,室温30分钟。 |
| 0.4%xSSPE,0.5%SDS,总体积200ml,60℃,30分钟。 | |
| 将膜包入塑料袋,在暗室贴在X光底片上。-70℃,自显影1-2天。 | |
| 冲片显影,观察结果(背景不清晰可重新洗膜再显影)。 |
(三)Northern blot
用于RNA分析,电泳条件与转膜方法与Southern blot不同外,RNA不必变性与中和,电泳时加电醛防止RNA发夹结构形成。其它步骤相同。
为了防止RNase水解需分析的mRNA,尽可能将器皿在160-180℃干热灭菌8小时以上,也可加0.1%焦碳酸二乙酯(DEPC)泡2小时,灭菌水淋洗干净,100℃干烤15分钟。不能干热灭菌的试剂等,也可加1%DEPC处理12小时(但不能用于Tris)后,100℃。加热15分钟(或高压灭菌15分钟)以抑制、分解RNase。1.0g琼脂糖,灭菌水80ml加热溶解。加x50TAE2ml,EB25μl,灭菌水至1000ml。样品缓冲液,x50TAE20μl,50%去离子甲酰胺500μl,甲醛180μl,混匀。约10-30μg/10μl纯化的RNA样品,加二倍(20μl)样品缓冲液(可点样一个凝胶孔lane)。60℃水浴15min,马上置冰浴。加1/10电泳指示剂。点样电泳,75-80V电泳4-5小时(指示剂泳动7~8cm),结束电泳。电泳期间正负极缓冲液要不断交换(可用蠕动泵),以免pH改变。电泳结束,将凝胶放在紫外光下可观察到人rRNA大亚基28S(5.1kb),小亚基18S(2.0kb),记下大小亚基移动距离(或拍照),可作为被测mRNA分子量分析的参照物。凝胶在50mmol/l NaOH中变性30分钟(低浓度碱,此步可省)。膜在10xSSPE中浸20分钟。用10xSSPE转移过夜(同Southern blot)。80℃1-2小时干燥。用探针杂交后,显影或显色。
(四)原位杂交(insituhybridization)
在保持细胞形状条件下,进行细胞内杂交,显影或显色。用于DNA或RNA分析。
细胞用离心涂片机涂片或组织切片置于载玻片上。4%多聚甲醛(PFA)/PBS固定(固定时间因标本而异10-20分钟,也可用含2%甲醛,0.05%戊二醛,2.5mmol/lCaCl2的0.1mol/L磷酸缓冲液pH7.3,500W微波炉中照射10-20秒,固定)。马上用PBS洗标本三次,2.5μg/ml蛋白酶K,37℃,5-20分钟(因标本和固定条件而定)处理。磷酸盐类缓冲液(PBs NaCl 137mmol/l,KCl 2.7mmol/L,Na2HPO48.1mmol/L,KH2PO41.5mmol/L)洗涤。4%PFA/PBS后固定,PBS洗一次,0.2%甘氨酸/PBS洗二次,每次15分钟。预杂交:42℃,1小时。预杂交缓冲液:10%硫酸葡聚糖,10%Denhardt液,0.5%吐温-20,250μg/ml鲑鱼精子DNA,500μg/ml酵母tRNA。杂交:42℃,4小时。用2x杂交缓冲液(4xSSC,0.2mol/L磷酸钠pH6.5,2xDenhardt)溶解已标记探针,使其终浓度为0.1-1.0μg/ml。DNA检测时把探针覆盖在标本上,置100℃5分钟(变性)取出,杂交。mRNA检测则先把探针置95℃水浴3分钟,立即置冰水浴,再覆盖标本上进行杂交。覆盖标本用液量100-200μl洗涤:2xSSC,50℃过夜。0.2xSSC,室温1小时。载玻片浸在缓冲液中。显色(试剂、方法参照斑点杂交的封闭-显色部分)20分钟-2小时,显微镜下观察结果。
二、核酸扩增技术
通过大肠埃希菌繁殖而增殖重组质粒,也是扩增核酸的手段。但在试管中有效扩增DNA的方法,是在90年代才开展起来的多聚酶链反应(polymerase chain reaction,PCR)新技术。由于PCR灵敏度高、特异性好、操作方便,在我国发展很快。目前,PCR技术结合分子生物学实验技术(如逆转录反应杂交技术等)开发了许多PCR新技术(reverse transcriptase PCR;PCR-single strandconformation polymorphism,PCR-SSCP;巢式PCR(二次PCR);热启动PCR等)。RT-PCR用于RNA分析;PCR-SSCP分辨率高,可用于点突变分析;二次PCR特异性好,灵敏度高;热启动PCR可提高特异性。
PCR技术原理是在了解DNA一级结构基础上设计引物(primer或sense,人工合成),DNA多聚酶可识别并结合引物,以单链DNA为模板,dNTP为底物,合成其互补链。通过反复变性,反复合成互补链的过程,达到DNA扩增的目的。人的DNA扩增引物长度多为20个核甘酸。(图18-7)

图18-7 PCR技术原理
PCR反应:DNA样品0.1-1μg,引物1,2各25-100pmol,dNTP各200μmol/L,Tris-HCl(pH8.3)10mmol/L,KCl 50mmol/L,MgCl21.5mmol/L,gelatin(明胶)0.01%,Taq多聚酶2.5-5U,矿物油一滴。93℃7分钟→50-55℃2分钟→70-72℃3-1.5分钟→93℃2分钟。

将反应物和Marker点样于凝胶一起电泳,紫外光下观察结果,拍照保存结果。反应物也可用于印迹(杂交)实验、多态分析、探针制备等。
注意事项:①DNA样品纯度高,Taq酶(和Klenow酶)有逆转录酶活性,DNA和RNA不能混在一起。②设计的引物分子内(特别3′端)和引物,1,2分子间不形成双链。选择性好,不增幅目的DNA以外区域的DNA。引物的G+C含量为40%-60%。③dNTP浓度过高易使Taq酶合成错误,一般在200μmol/L,有人建议40-50μmol/L。④KCl>75mmol/L对酶有抑制作用。Mg2+浓度过高,NDA不易变性,>10mmol/L可抑制酶活性40%-50%。⑤多聚酶:Taq多聚酶从嗜热菌分离得到(1989年,在此以前1985年利用Klenow酶因不耐热每经过一次热变性要补加一次酶),现在经基因克隆的重组体产物Taq酶最适pH8.3~8.8(室温8.3-8.4),反应温度75-80℃,95℃以上失活明显。无3′→5′校读活性,对SDS敏感(<0.01%活性也受到抑制,加吐温-20可抵制SDS抑制)。该酶有逆转录酶活性。⑥退火温度一般设定为Tm-5℃,但实际上与引物一级结构序列有关,设计不当可造成非特异性退火,而造成非特异扩增,此时应调整温度和相应的时间。⑦循环次数受到的影响因素较多,增加次数可提高灵敏度,但易出现非特异扩增。⑧PCR灵敏高,被检样品极易被污染,样品纯化,扩增,检测区域应分开。房间应经常用紫外光照射。扩增物应定点丢弃,处理。
三、核酸限制性片段长度多态性(RFLP)分析
RFLP分析是限制性内切酶、核酸电泳、印迹(blot)技术、探针-杂交技术的综合应用,多用于临床遗传性疾病的基因诊断。基本原理是遗传性疾病多有基因的缺陷,如基因缺失、基因插入、编码区小范围核苷酸序列改变或点突变等。前二者可将纯化的正常人和病人DNA同时用限制性内切酶切成片段,经电泳分离、印迹、探针杂交等找到目的基因。由于患者基因的缺失或插入,造成被限制性内切酶切开的片段(分子量)大小与正常人发生差异(限制性片段长度多态),使其电泳迁移率发生差异,而达到基因诊断的目的。小区域或点突变,可选用一个限制性内切酶的切点正好在这一变异位置,使切割作用和正常人发生差异(如正常人基因可切断而患者不能,或反之),达到诊断目的。现在PCR产物经变性为DNA单链,用聚丙烯酰胺凝胶电泳分离,只要有一个核苷酸发生变异,其电泳迁移率就会改变,进行多态分析(PCR-SSCP)。实际上把基因的异常位置设计在PCR扩增区域内,就能进行多态分析。(图18-8)
核酸纯化→内切酶作用→电泳分离→Southern blot→探针-杂交→显影,显色
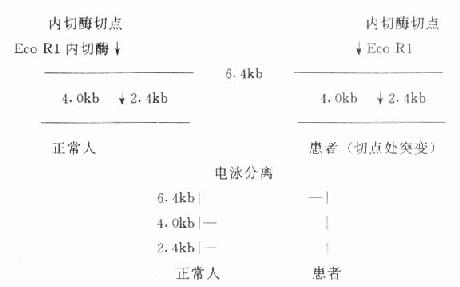
图18-8 点突变Eco R1RFL
四、核酸一级结构(核苷酸序列sequence)分析
核酸一级结构分析是基因分析最直接的第一手资料。目前DNA、RNA以及蛋白质的一级结构分析中,DNA的碱基序列分析方法多样、简单、快速。下面就M13噬菌体-双脱氧核苷酸(ddNTP)的DNA测序法介绍如下。
M13是单链DNA噬菌体,转染宿主菌体复制为双链增殖,又以单链形式透出菌体。把待测DNA重组插入双链M13复制型DNA中,经培养扩增,可分离得到单链M13DNA(含待测单链DNA的碱基序列),即复制模板。在待测DNA的3′端人工合成引物,在DNA聚合酶Ⅰ作用下,复制链延长。在ddNTP(2′,3′双脱氧核糖核苷三磷酸)存在下,复制延长随机受阻,形成分子量(长度)大小不等的片段。电泳分离,自显影,读图18-9分析碱基序列。

图18-9 核酸碱基序列分析
0.2-0.3mm厚度聚丙烯酰胺-尿素凝胶,高压(1600V)电泳,可分辨一个核苷酸的差异。电泳后干燥凝胶,自显影,自下往上读图,得到5′→3′的合成链碱基序列,其互补链是待测DNA的3′→5′碱基序列。
-
《临床生物化学》 中的相关章节:
……
第三节 酶活性测定条件的选择和限定
第四节 测定酶活性浓度的两大类方法
第十八章 诊断分子生物学基本技术
第一节 概述
第二节 分子生物学实验基础
第三节 分子生物学实验诊断技术(当前页)
第四节 诊断分子生物学技术的临床应用
第十九章 临床生物化学分析仪的性能与应用
第一节 临床生化自动分析仪的类型
第二节 临床生化自动分析仪的性能评价与合理选用
……